
必揚醫藥(Beyang Therapeutics),成立于2021年,是一家既年輕又成熟的Biotech公司。基于ExCEED(Experiment + Computation w/ Enhanced Evaluation & DMPK)技術平臺,必揚專注于計算模擬技術和藥物發現實驗技術的高度融合,尋找更快、更智能的方法來發現新的和更好的藥物。
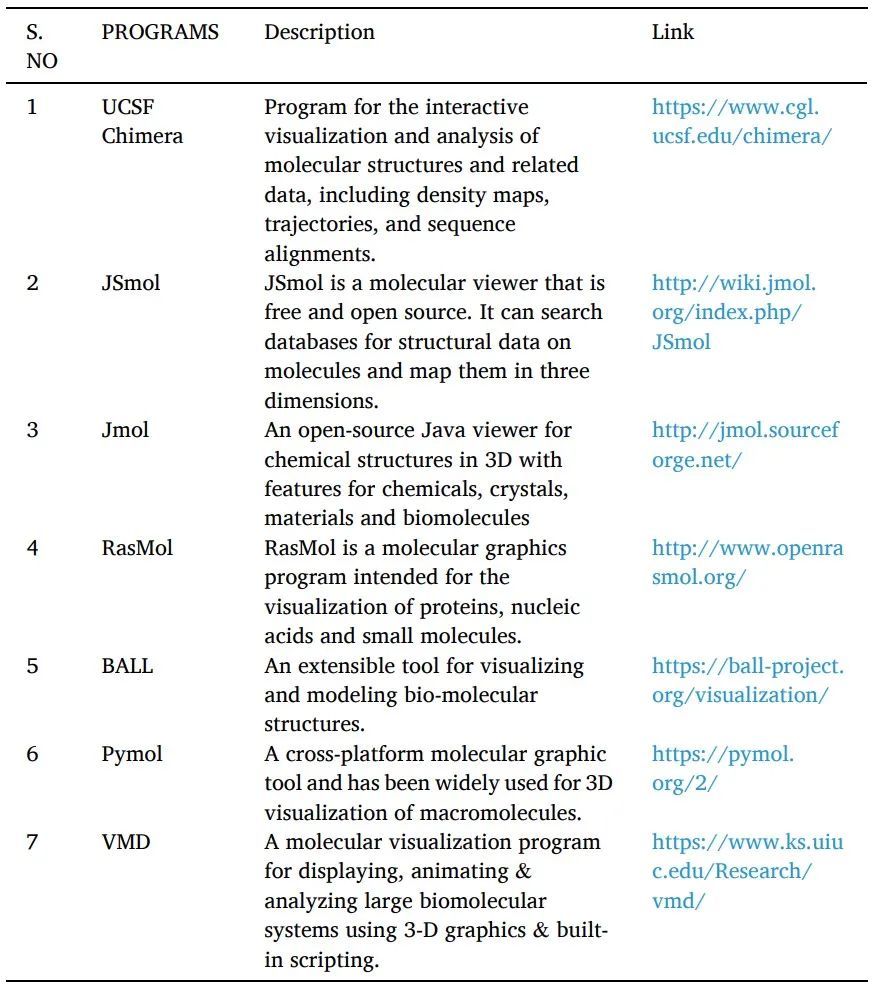
基于多年在行業深耕的經驗和獨特的創新視角,胡齊悅在2021年創立了必揚醫藥。他的初衷是:“我希望必揚作為創新藥物的‘夢工廠’,以另外一種方式,匯聚有各自專長的人組成精煉小團隊,研發出Best in Class的分子,加速藥物研發進程。”從分子設計、成藥性評價與生物學評價三個核心環節入手,必揚期望打造一個能不斷產出高質量分子的創新藥“夢工廠”,助力中國創新藥走出國門、邁向海外。

01
計算機土壤上的
創新藥“夢工廠”
全球高手組隊打造“夢工廠”,專注創新藥
必揚的團隊來自世界各地,精煉且資深。
胡齊悅在Pfizer(輝瑞)La Jolla實驗室工作十多年后,選擇回國加入恒瑞醫藥。在中國創新藥的發展萌芽階段,繼續深耕近十年后,成立了必揚醫藥。公司創始團隊成員均擁有超過20年新藥研發與計算模擬結合的實踐經驗,累計對20余個臨床階段候選藥物或已上市新藥的研究與開發做出過重要貢獻,例如Adebrelimab、Axitinib、Crizotinib、Sunitinib、Sylatron等等。
必揚的“夢工廠”模式不僅在于小而精的團隊本身,還在其強大的創造能力。
必揚的優勢體現在,具有挑戰性的靶點與適應癥選擇,以及臨床前項目管線的推進和有明顯差異化的數據。
成立不到三年,已經有一款以滴眼治療眼底疾病的項目管線正在推進臨床注冊申報研究,該適應癥臨床可用藥物匱乏,且無治療指南推薦藥物,未滿足臨床需求巨大。同時,另一款治療高危型白血病復發難治亞型的項目也即將進入臨床注冊申報研究,該適應癥標準療法為化療,尚無針對性靶向療法獲批,亦存在非常明確的未滿足臨床需求。

無論是眼科項目還是腫瘤項目,均在臨床前研究中,通過與所選適應癥/靶點上最好的藥物頭對頭比較,充分證明其 Best in Class 潛力。
出生自帶計算機技術基因:CADD+AIDD
三年前,速石寫過一篇文章,盤點了全球44家頂尖藥企用人工智能輔助新藥研發的現狀。到今天,依然可以得出這個結論:我們離主要靠計算機發現藥物,通過一大堆機器人完成實驗測試,然后將它們送給患者的世界還相距甚遠。
但這并不意味著,我們不能繼續往前。
正是計算機技術的應用讓創新藥研發這件事情變得扁平化,把中國與世界的距離拉近。
必揚從誕生的一刻起就自帶計算機技術基因 (CADD+AIDD)。
胡齊悅在輝瑞就專注于計算機輔助藥物研發的工作,在恒瑞任職期間,帶領團隊充分發揮計算模擬與實驗的協同作用,并將計算模擬的應用從小分子領域進一步拓展到包括抗體、重組蛋白、ADC和RNA等生物大分子形式。

胡齊悅表示,相比Me Too,做Best in Class會更需要計算機技術的幫助,或者說,借助這樣的技術可以把這件事做得更好。比如分子設計,一般靠的是研究人員經驗,他知道什么樣的化學結構可以跟靶點有很好的結合。但一個人即使再有經驗,也不可能窮舉所有可能性,這時候可以借助計算機的力量把已知可能的分子都設計出來,通過一個評價體系來排序并選擇。
而一個奇思妙想的結構,有時能點燃創造的火花。
而像成藥性評價,如果通過AI機器學習的算法模擬實驗數據訓練預測模型可以節省人力、物力以及實驗資源,并能更快獲得最終的實驗結果。
02
專注核心
借助外部的力量
Biotech的主業,始終是前沿靶點和技術,解決未滿足臨床需求。必揚的“夢工廠”模式,最重要的就是專注于自己的核心目標,同時充分借助外部的力量。
隨著整個制藥行業的發展與成熟,CRO、CMO、第三方研究所等專業機構的出現,為行業的發展帶來了全新的商業模式,很多非核心業務都可以被拆分出來,實現“分布式”分工協同,讓必揚的發展更輕松與高效。
比如,做新藥研發云平臺的速石科技就是必揚的合作伙伴之一。
過去一年,必揚在速石平臺的使用趨勢如下圖:

最高達到1500+核心并發,最低值是0。
真正做到:需要的時候就在那。無需預測,想用就用,用完即走。
給必揚帶來的好處在于:
首先,輕松無負擔。無需增加自有資產,占用公司現金流;
其次,使用方式靈活。需要的時候購買,用完即走,不需要培養一個專門的團隊;第三,減少前期摸索時間與降低試錯成本。
還在美國工作的時候,胡齊悅就已經知道云的優勢了。
在恒瑞時他曾經也想上云,但即便在大型藥企,從無到有地搭建一套可用研發環境依舊是復雜的:從爭取公司資源,到招聘專業人才,包括后續一系列采購設備、設計底層架構、打通網絡、部署系統、管理維護集群等工作。
考慮到這些繁瑣耗時的工作都需要他付出相當大的精力,就一直沒往這個方向投入。
而在本地機器購買完成的那一刻起,就開始了固定資產的貶值。

創立了必揚后,作為一家企業的創始人,面臨的問題就更多更現實了。
專門準備一個機房,招一堆人來搭建整個環境和底層架構,再專門找IT維護。他直接表示:“這個對于多數Biotech不實際”。而且每年可以投入到機房的費用是一定的,但是需要做計算的事其實是在增長的,這種模式也非常不靈活,很難匹配一家高速增長的企業需求。
在胡齊悅看來,速石提供的新藥研發云平臺是一條捷徑或者說高速路,而這對藥物研發企業具有很重要的價值。

面對多樣的藥物研發場景和BIO/AI工具,如分子對接、虛擬篩選、蛋白質折疊等,速石能為像必揚這樣的用戶提供三大核心價值:
1. 標準藥物研發環境的規劃與搭建。
速石站在醫藥公司整個新藥研發體系與架構視角進行標準藥物研發環境的規劃與搭建,為企業提供一整套即開即用的CADD/AIDD研發平臺,一體化產品端到端交付到用戶手里。
有效降低用戶的前期投入與摸索成本,任何起步的公司都可以嘗試,沒有使用門檻,且對企業的未來擴張有極強的兼容性與彈性。
2. 藥物設計研發流程的構建與驗證,BIO/AI應用的適配和銜接。
速石深入藥物研發業務場景,為用戶提供藥物研發流程自動化的構建與驗證,對上層BIO/AI應用運行提供支持與優化,同時與底層資源聯動,給用戶更靈活、更高效使用資源的能力。
一方面把用戶原來手動做的工作自動化,大大提升用戶的工作效率;另一方面為用戶提供應用與資源層面的最佳實踐參考,解決了研發場景里普遍存在的資源利用率不高、成本難以把控等問題。
3. 處理研發與IT領域的交叉問題。
做計算機輔助藥物研發,遇到的通常是介于藥物研發與計算機IT領域的交叉問題,定位困難,解決復雜,非常消耗用戶精力。速石能幫用戶預判部分問題,通過自動化減少部分問題發生,定位并解決部分問題,幫助用戶專注更重要的藥物研發工作。

此外,速石的新藥研發平臺還能支持市面上所有主流新藥研發應用,和AlphaFold、RoseTTAFold等常用AI框架的快速搭建,并提供開源/自有分子庫構建與治理,如Zinc、Drugbank、Maybridge、Enamine等等。
速石常見BIO應用實證:
Vina/ Amber/ MOE/ LeDock
03
思路拓展:
一個假設性問題
我們問了胡齊悅一個假設性的問題:
“如果沒有預算,你會怎么使用我們平臺?”
或者換一種表達方式:“如果沒有資源限制,你會如何使用計算機技術來輔助藥物研發?”
他回答:“會用得更多。”
多跟少之間的權衡點具體有哪些?
第一, 研究的全面性。
面對資源限制,企業首要考慮的是該研究項目的必要性與重要性,然后計算需要投入的時間與資源量。這可能會嚴重影響研究的全面性。
以設計分子為例,藥物研發人員會從現有的資源與認知出發,制定研究計劃,無法考慮超出資源之外及認知之外的可能性。
隨著研究邊界的不斷拓展,可能會因為有限的資源與認知,對某一個項目帶來致命的、決定性的負面影響。而研發人員永遠無法知道錯過了什么。

第二,應用與資源的適配。
資源限制會對研發人員使用應用的方式提出要求,他們需要能根據任務特性挑選更合適、性價比更高的機器。
如果只選用大機型執行任務,將會浪費大量資源,而如果錯誤選擇了小機型,很可能中途會面臨任務失敗。
如何找到應用與資源類型、資源規模的最佳適配,需要不斷地學習和實踐。
第三,時間與金錢的取舍。
從事藥物研發,研發速度不是唯一的考量。要提升研發的深度與廣度,實現整體效率的提升,充足的資源必不可少,但這也意味著企業需要付出更多的成本。企業需要在效率與成本間做抉擇,甚至需要根據外部環境的變化做動態調整。這也是大部分企業都在面臨的難題。
Take Away
- 高手組團,小而精且靈活,計算機技術加持,專注有挑戰性的靶點與適應癥,我們有理由期待必揚醫藥將在創新藥研發這條路上越走越遠。畢竟“夢工廠”是為大家制造夢想的地方,而生命/健康是人類永恒的追求。
- 對計算機技術輔助藥物研發的信心,也就是CADD/AIDD到底能在創新藥研發過程中起到多大的作用,必揚作為一家Biotech給國內的醫藥公司們成功打了個樣。而我們提供給必揚以及創新藥公司的價值,就是讓他們盡可能專注自己的研發業務,其他事情都能交給我們。
- 生物制藥這個行業,05、06年開始一直往上走,到2015年達到頂峰,直到2021年開始往下走了。但中國醫藥從“仿制藥”轉型“創新藥”這個大方向是不會變的,只是泡沫退去之后,不再有冒險和神話,只留下專業、理性和冷靜做事的人和公司。
關于必揚
蘇州必揚醫藥科技有限公司是一家創新的制藥公司,利用其專有的ExCEED 平臺來加速針對未滿足臨床需求的新藥發現和開發進程。創始人和管理團隊平均擁有超過20年新藥研發經驗,曾任職于輝瑞、默克、阿斯利康、先靈葆雅和恒瑞。他們在藥物靶點選擇、CADD、AIDD、生物篩選和優化以及DMPK方面具有豐富的經驗。他們共同參與促成了20 多個臨床候選分子的開發,其中多個藥物(Adebrelimab、Axitinib、Crizotinib、Sunitinib、Sylatron等)已獲得 FDA 和 NMPA 的批準,于中美兩地上市。
必揚建立了專有的計算機加速藥物發現整合平臺ExCEED,該平臺利用新的分子生成和優化方法 MMPT 和計算機 DMPK 模擬預測來指導臨床前候選分子的發現和開發,能夠大幅拓展可探索化學空間,打破制藥專家固有思維的桎梏,從而提高藥物研發效率,獲得更優質的分子。公司專注于眼科、腫瘤等具備臨床未滿足需求的疾病領域,旨在打造全球新藥研發界的“DreamWorks”(夢工廠),充分發揮團隊在藥物發現階段的優勢,自建管線與合作授權開發(License-out or Co-development) 并行。
關于fastone云平臺在各種BIO應用上的表現,可以點擊以下應用名稱查看:
速石科技新藥研發行業白皮書,可以戳下方查看:
-END-
我們有個一站式新藥研發平臺
集成行業應用與自編譯軟件
支持AlphaFold、RoseTTAFold等常用AI應用
可視化Workflow隨心創建、便捷分享
提供Zinc、Drugbank等開源/自有分子庫
CADD專家團隊全面支持
掃碼免費試用,送200元體驗金,入股不虧~

更多BIO電子書歡
迎掃碼關注小F(ID:iamfastone)獲取

你也許想了解具體的落地場景:
王者帶飛LeDock!開箱即用&一鍵定位分子庫+全流程自動化3.5小時完成20萬分子對接
1分鐘告訴你用MOE模擬200000個分子要花多少錢
155個GPU!多云場景下的 Amber自由能計算
提速2920倍!用AutoDock Vina對接2800萬個分子
新藥研發平臺:
國產調度器之光——Fsched到底有多能打?
創新藥研發九死一生,CADD/AIDD是答案嗎?
全球44家頂尖藥企AI輔助藥研行動白皮書
近期重大事件:
速石科技入駐粵港澳大灣區算力調度平臺,參與建設數算用一體化發展新范式
速石科技成NEXT PARK產業合伙人,共同打造全球領先的新興產業集群
速石科技出席ICCAD2023,新一代芯片研發平臺助力半導體企業縮短研發周期
速石科技與芯啟源開啟戰略合作,聯手打造軟硬件一體芯片研發云平臺
速石科技作為特邀服務商入駐IC PARK,合力打造集成電路產業新生態
速石科技成為國家“芯火”深圳雙創基地(平臺)戰略合作伙伴,推動國產EDA公共技術服務云平臺建設


本篇來說下CADD對藥物設計領域產生了重大影響。此外,CADD與人工智能(AI)、機器學習(ML)和深度學習(DL)技術相結合,處理大量生物數據,減少了與藥物開發過程相關的時間和成本。
1.概述
計算機輔助藥物設計(CADD)結合了各種計算機工具,以識別和開發有前途的lead。CADD包括計算化學、分子建模、分子設計和合理的藥物設計。在當今的大數據環境中,訪問大量數據并不能保證獲得適用的預測模型。為了預測治療效果和副作用,必須開發系統地解決大量、多維和稀疏數據源的技術。
1.1 計算機輔助藥物設計
CADD基于蛋白質或配體的3D結構的可用性,利用兩種不同的技術進行藥物發現:基于結構的藥物設計(SBDD)和基于配體的藥物設計(LBDD)(圖1)。在某些情況下,兩種技術的整合在查找先導分子方面顯示出良好的準確性。
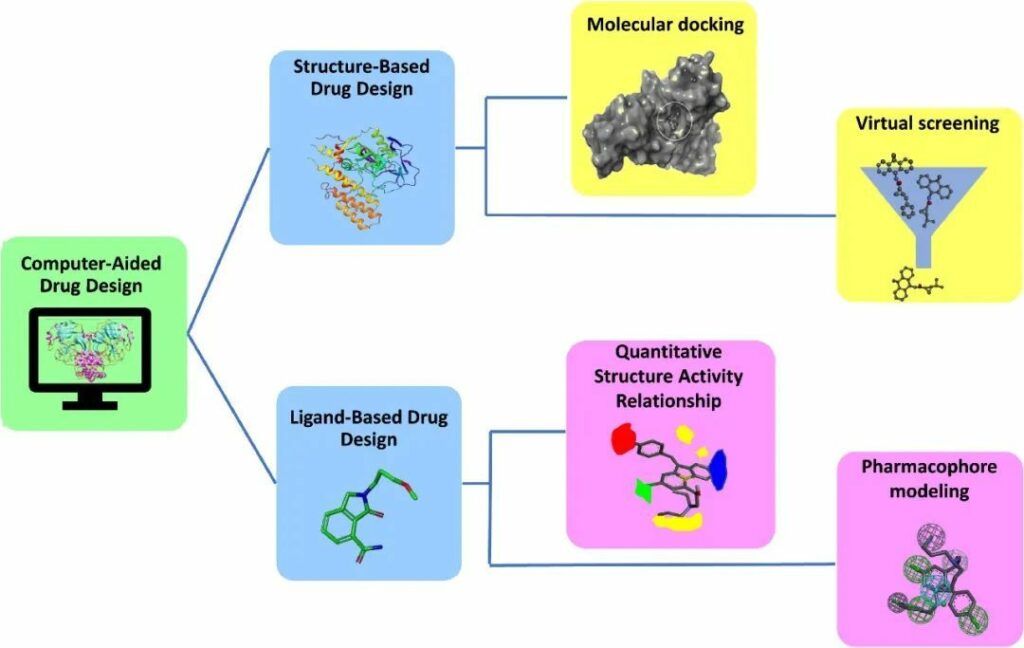
圖1 -CADD概覽
1.1.1 SBDD
基于結構的藥物設計(SBDD):隨著越來越多生物分子的三維結構的公開,SBDD在藥物發現和設計方面的新時代已經開始。SBDD已成為制藥行業中生成和優化配體的可能手段。靶標的識別、結合位點的鑒定、分子對接、虛擬篩選和分子動力學是SBDD的基本步驟。
1.1.1.1靶標準備
準備靶點大分子結構是SBDD中最關鍵的一步。由于X射線和NMR結構解析技術的快速發展,沉積在蛋白質數據庫(PDB)中的蛋白質的3D結構很容易獲得。當目標蛋白的三維結構不可用時,計算方法,如比較或同源建模(comparative or homology modeling)、threading和ab initio已經能成功地從蛋白質的序列中確定其結構。
同源建模或比較建模:可以使用各種計算結構預測技術(包括同源性建模)從其氨基酸序列推斷蛋白質3D結構(表1)。它被認為是精度最高的計算結構預測方法。其中有幾個簡單且易于遵循的步驟:尋找具有相似序列的結構模板蛋白,對齊它們的序列,使用對齊的區域坐標,預測目標缺失的原子坐標,模型構建和細化。NCBI基本局部比對搜索工具(BLAST)是用于序列相似性搜索的最廣泛使用的生物信息學序列比對工具之一。
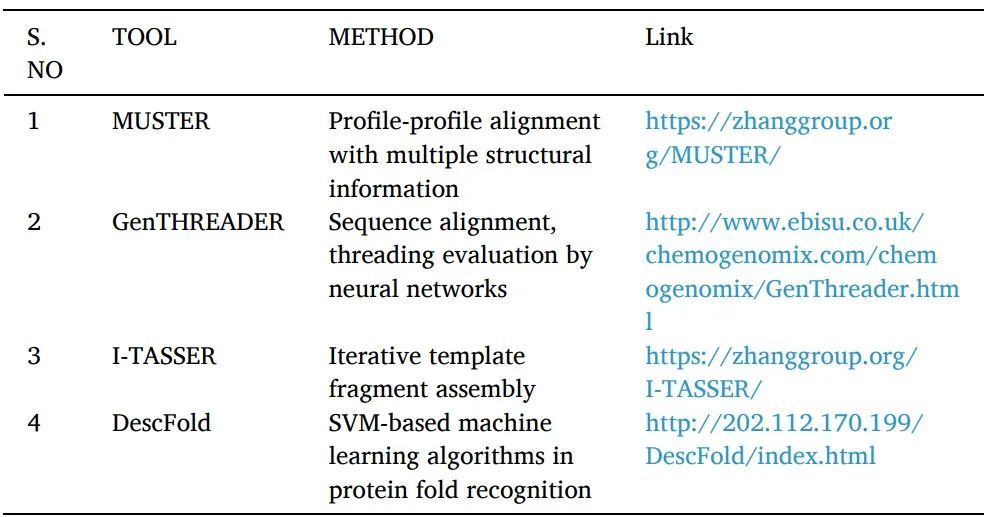
Fold recognition or threading:用于尋找具有可比折疊但沒有序列相似性的蛋白質。一個已知的蛋白質結構的序列被感興趣的目標的查詢序列所取代,對于該結構是未知的。然后用各種評分系統對產生的"threaded"結構進行評估。對每個數據庫的經驗確定的三維結構重復這一過程,提供與查詢序列最匹配的結構(表2)。它被運用于SBDD研究中。
表2 threading方法工具
從頭或從頭建模:當結構中沒有足夠的同質性來進行比較建模時,將執行從頭或從頭建模(表3)。
表3 用于從頭建模的工具

1.1.1.2活性結合位點的鑒定和表征
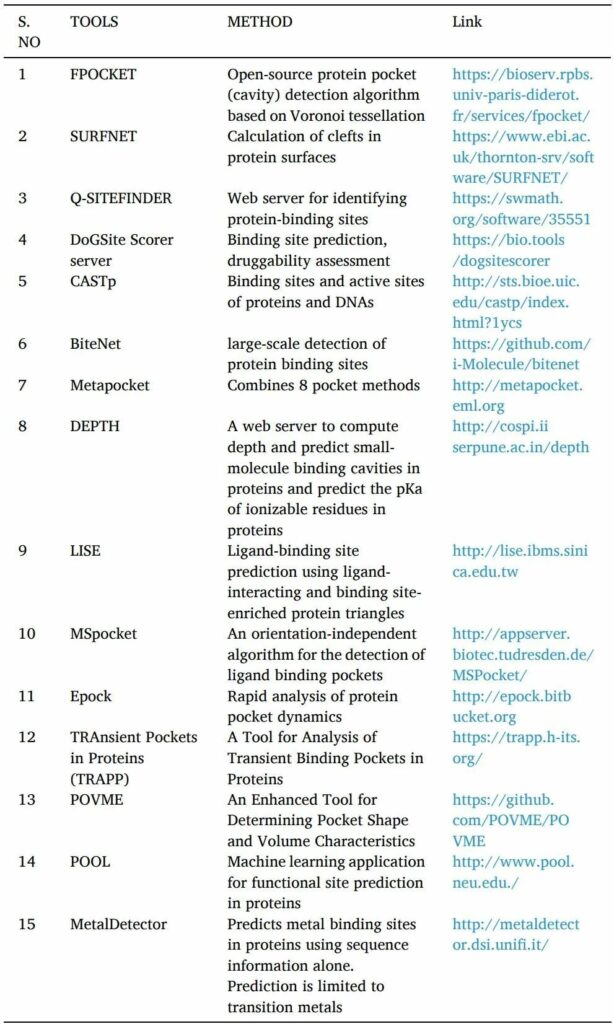
藥物活性需要蛋白質和配體的相互作用。只有找到高親和力結合位點才有可能。在基于結構的藥物發現方法中開發新方法在很大程度上取決于識別靶蛋白上的可藥腔或口袋。
POCKET,SURFNET,Q-SITE FINDER,DoGSite Scorer server,CASTp,NSiteMatch,metapocket等工具是用于預測靶點蛋白結合位點的計算機工具。
找到結合位點后,使用諸如Epock,TRAPP和POVME等工具或服務器來確定結合口袋的體積(表4)。
1.1.1.3 分子對接
表4 結合位點預測工具
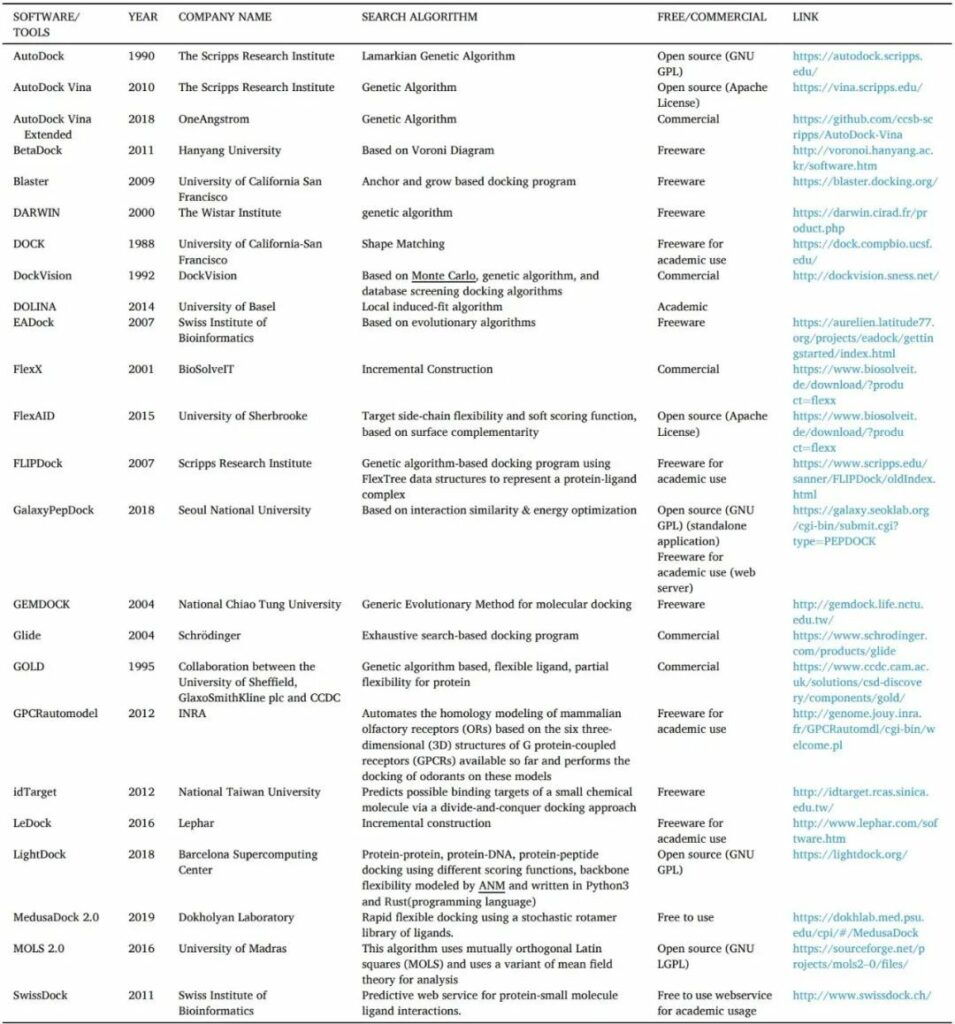
分子對接是一種用于確定配體分子在大分子靶標結合位點中的構象和取向(統稱為“位置”)的技術。
搜索算法用于生成姿勢,然后使用評分技術進行排名。許多生物過程,如信號傳遞、細胞控制和其他大分子組裝,依賴于分子識別,如酶-底物、藥物-蛋白質、藥物-核酸、蛋白質-核酸和蛋白質-蛋白質相互作用。采樣和評分是蛋白質-配體對接方法的兩個關鍵組成部分。
配體采樣和蛋白質靈活性是采樣的兩個方面,指的是在蛋白質結合位點附近創建可能的配體結合方向/構象。評分使用物理方法或經驗預測單個配體取向/構象的結合緊密性。
表5 對接工具/軟件的清單
蛋白質-配體對接的分類方法有幾種。
根據蛋白質靈活性,方法分為四類:
軟對接:在對接模擬中,它放松了原子間Vander Waals接觸,允許配體和蛋白質之間略有重疊。
側鏈靈活性:早期的研究之一是Leach的配體對接方法,該方法使用旋轉體庫來結合離散的側鏈靈活性。從那時起,已經提出了一系列新技術,用于在配體對接中添加連續或離散的側鏈靈活性。
分子弛豫:第三種方法考慮蛋白質的柔韌性,通過使用剛體對接將配體引入結合位點,然后松弛蛋白質主鏈和附近的側鏈原子。初始剛體對接允許蛋白質和插入的配體取向/構象之間的原子沖突,以適應蛋白質構象差異。使用蒙特卡羅(MC)模擬,分子動力學模擬或其他方法松弛或最小化復合物。
蛋白質集合對接:添加蛋白質柔韌性的最廣泛使用的方法涉及蛋白質結構的集合,以反映各種構象變化。
根據配體采樣,方法分為兩類:
有兩種配體采樣算法:系統搜索和隨機算法(表6)。
表6使用隨機或系統搜索算法的軟件/工具
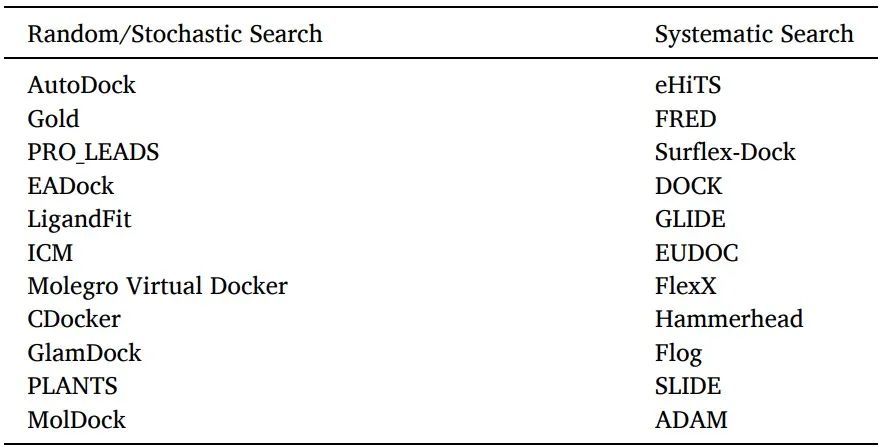
系統搜索:對于靈活的配體對接,通常使用系統搜索技術,通過探索配體的所有自由度來創建所有潛在的配體結合構象。
隨機算法:通過在構象空間和配體的平移/旋轉空間的每一步對配體進行隨機修改,在隨機算法中對配體結合取向和構象進行采樣。
根據評分功能,方法分為三類:
對接分數是基于評估復合體能量親和力的評分函數的計算。這些評分函數可以在分子力學、經驗數據、專業知識或共識庫上找到。共識評分(Consensus scoring)是一種通過組合幾種評分算法的結果來預測化合物對特定靶標的結合親和力的方法。根據推導方法分為三個基本類別:基于力場、基于經驗和基于知識的評分函數。
力場(FF)評分函數:基于配體結合能分解為單個相互作用項,例如范德華(VDW)能量,靜電能,鍵拉伸/彎曲/扭轉能等,使用一組派生的力場參數,例如AMBER或CHARMM力場。
經驗評分函數:配合物的結合能得分是通過將幾個加權經驗能項(如VDW能,靜電能,氫鍵能,脫溶劑化項,熵項,疏水性項等)相加得出。
基于知識的評分函數:直接從實驗確定的蛋白質配體復合物中的結構信息生成平均力的電位,由玻爾茲曼反比關系描述為基于知識的評分函數提供基礎。
1.1.1.4 虛擬篩選(VS)
在計算機中,從化學數據庫中選擇有希望的化合物的方法被稱為虛擬篩選,并且可以被認為是實驗生物學評估方法的計算機化等效物,如高通量篩選(HTS)。
VS分為兩類:(i)基于結構的虛擬篩選(SBVS)和(ii)基于配體的虛擬篩選(LBVS)(圖2)。

圖2 基于結構和基于配體的虛擬篩選概述
基于結構的虛擬篩選(SBVS):這是一種基于計算機的方法,用于在早期藥物開發項目中針對特定治療靶點搜索化合物庫中的新型生物活性化合物。SBVS中的化合物數據庫停靠在預定的靶標結合位點。除了預測結合模式外,SBVS還為對接的分子分配排名。該評級可以用作選擇有前途的分子的唯一標準,也可以與其他評估方法結合使用。進行實驗以確定所研究分子靶標上指示藥物的生物活性。SBVS包括四個步驟:(i)分子靶標準備(ii)化合物數據庫選擇(iii)分子對接和(iv)對接后分析。
表7 用于以圖形方式顯示SBVS和分子對接結果的程序
基于配體的虛擬篩選(LBVS):通過采用稱為基于配體的虛擬篩選的計算技術,可以根據有效結合到靶標的配體的信息生成靶蛋白的模型。之后,使用該模型預測新配體與靶標結合的可能性。LBVS是唯一沒有靶蛋白3D結構的方法。LBVS試圖使用已知的活性化學物質作為輸入信息來識別具有相似屬性的結構多樣化的分子。
1.1.1.5 分子動力學(MD)模擬
這種復雜的物理技術基于牛頓引導原子間相互作用的運動方程。它用于預測分子系統中每個原子相對于時間的位置。分子動力學(MD)模擬對于研究蛋白質行為至關重要。在MD模擬中,化學鍵和鍵角使用簡單的虛擬彈簧描繪,而二面角則使用正弦函數處理。GPU 最初設計用于加速視頻游戲,現在正用于顯著加速分子動力學模擬。分子力學泊松-玻爾茲曼表面積(MM / PBSA),線性相互作用能(LIE)(和自由能擾動方法(FEP)是用于自由能計算的一些MD應用,以關聯實驗和計算的小分子與蛋白質的結合親和力。分子動力學模擬可以使用牛頓物理學和力場(如Amber或CHARMM OPLS,GROMOS和粗粒度力場(表8)計算構象軌跡作為時間的函數。
表8 MD仿真中使用的力場列表

AMBER(具有能量細化的輔助模型構建):AMBER是一組分子模擬程序和一組用于模擬生物分子的分子機械力場。Peter Kollman的小組在1970年代后期在加州大學舊金山分校開發了這種方法,以研究各種分子,如蛋白質,DNA,RNA,碳水化合物,有機分子,蛋白質模擬物,脂質和氟化芳香族氨基酸。為了施加分子對稱性,在全原子原子輔助模型構建與能量細化(AMBER)系列力場中,部分電荷被分配具有靜電表面電位。
CHARMM(哈佛大學大分子力學化學):CHARMM程序最初由哈佛大學的Martin Karplus教授小組開發,該小組協調經驗力場參數化的努力。它具有針對各種分子參數化的特定力場。CHARMM力場中的部分電荷通常適合從頭計算的尺度能量。
OPLS(液體模擬的優化電位):與實驗結果相比,OPLS力場在預測蒙特卡羅模擬獲得的結構和熱力學特性方面表現出準確性。
OPLS-AA力場,用于重現小分子的量子力學構象能量分布。此外,它還從AMBER中獲得了幾個粘結參數。
GROMOS(格羅寧根分子模擬):GROMOS是一種用于研究生物分子系統的分子動力學的多用途計算機模擬工具。它還具有內置力場,包括蛋白質、核苷酸、糖和其他分子。它可用于模擬各種化學和物理系統,包括玻璃、液晶、聚合物、晶體和生物分子溶液。
粗粒度力場(CG):CG力場通過減少模型中的自由度數來降低計算的計算成本,從而允許對更大的系統進行更長時間的仿真。粗粒度(CG)模型有兩種常規方法:自下而上和自上而下。許多化合物的極性相和非極性相之間的自由能分配是CGMartini力場的基礎。Martini力場也是與原子模型密切合作開發的,特別是在束縛相互作用方面。
1.1.2 LBDD
在沒有關于受體的3D信息的情況下,可使用基于配體的藥物設計。該技術依賴于與感興趣的生物靶標結合的分子知識。
與靶標結合的已知配體的化學指紋圖譜用于分子相似性方法,以使用分子庫進行篩選來鑒定具有相似指紋的化合物(表9)。配體相似性搜索方法是有效的,因為結構相關的化合物具有相當的結合特性。
表9 小分子數據庫

QSAR和藥效團是LBDD的兩種方法。
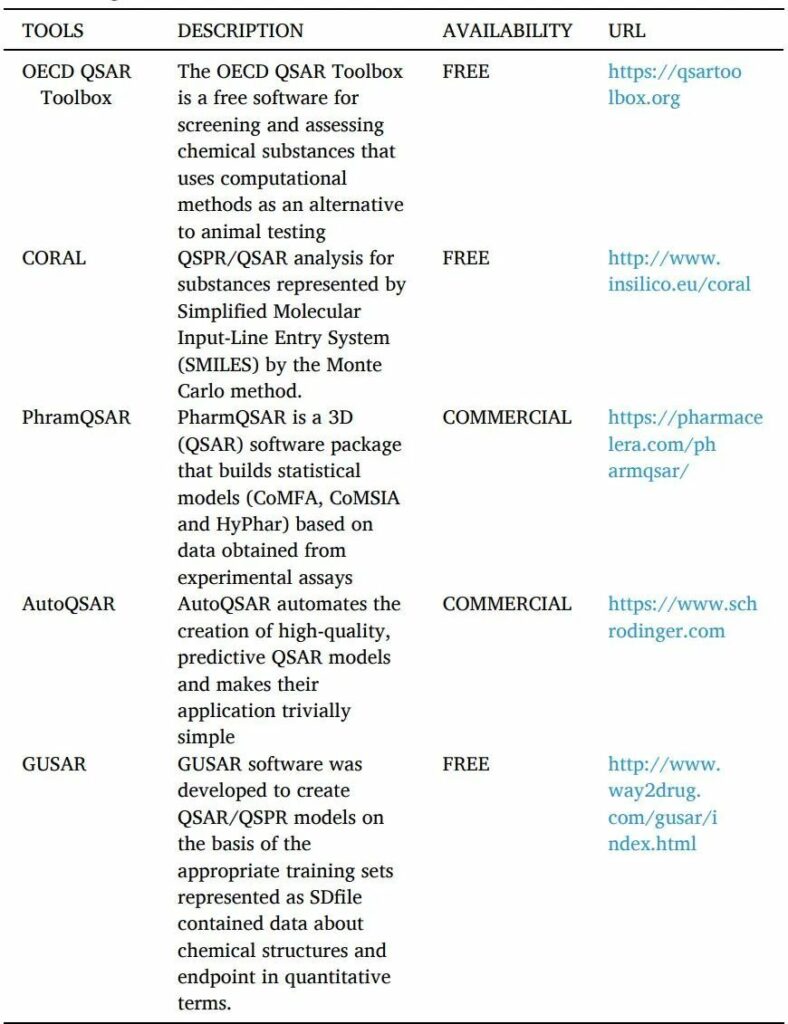
1.1.2.1 QSAR
QSAR是一種計算機化的統計工具,用于解釋觀察到的由替換產生的結構變化(表10)。這些模型用數學方法證明配體的結構特性如何影響與之結合的靶點的活性反應。可用于建立QSAR模型的分子參數可能包括電子、疏水、立體和亞結構效應。
QSAR的工具如下。
表10 QSAR工具
詳細參數:
電子效果:電離常數、Σ取代基常數、分布常數、共振效應、場效應、分子軌道指數、原子/電子凈電荷、親核超離域性、親電超離域性、自由基超離域性、最低空分子軌道和最高占據分子軌道的能量、前沿原子-原子極化率、分子間庫侖相互作用能、由一組電荷在點(A)處產生的電場分子。
疏水參數:分配系數、Pi取代基常數、液-液色譜中的Rm值、高壓液相色譜(HPLC)中的洗脫時間、溶解度、溶劑分配系數。
空間效應:分子內空間位阻效應、空間位阻取代基常數、超共軛校正、摩爾體積、摩爾折射率、MR 取代基常數、分子量、范德華半徑原子間距離。
子結構效應:三維幾何碎片和分子性質。
QSAR的步驟(圖3)如下:

制備用于QSAR實驗的分子:獲得一組在類似生物學測定中測試并顯示出廣泛作用的同屬配體。
訓練集中描述符的選擇:識別并確定與化合物理化性質相關的分子描述符。
計算訓練集中描述符的值:將分子隨機分為兩組:訓練集和測試集。使用訓練集,識別并計算可以解釋描述符值與生物活性之間關系的相關系數。
內部和外部驗證評估:使用測試集分子,評估統計方法的穩定性。
1.1.2.2 藥效團建模
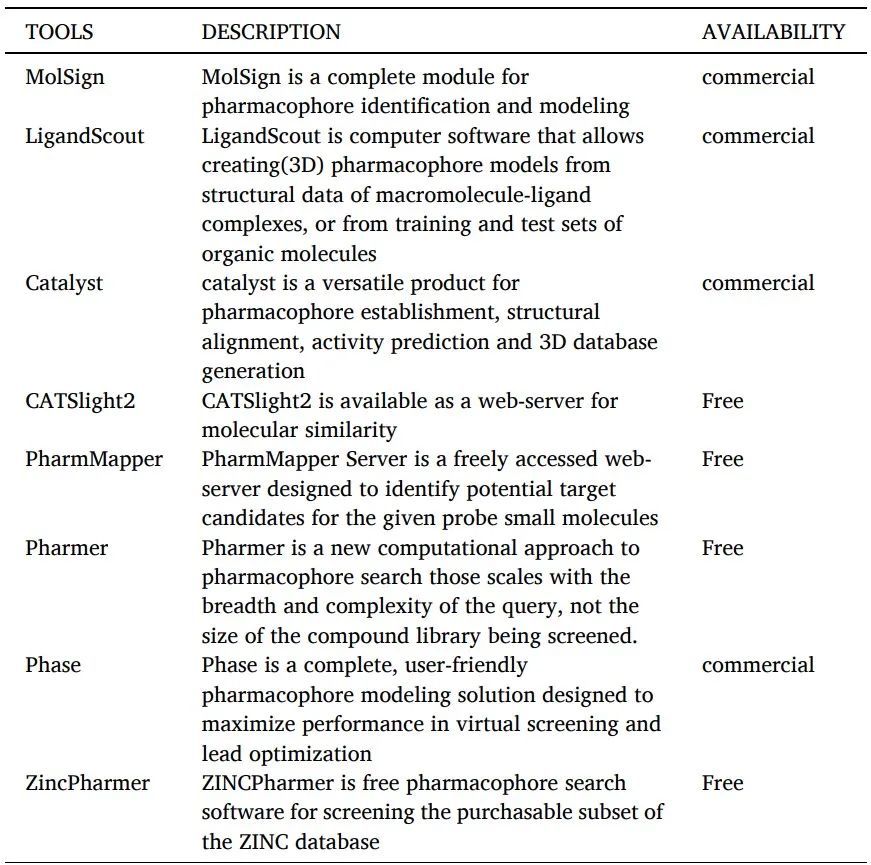
根據IUPAC的定義,藥效團是“實現與給定生物靶標的最佳超分子相互作用并觸發或預防其生物反應所需的空間位阻和電子特性的集合”。藥效團是對生物大分子識別配體所需結構特性的抽象描述(表11)。
藥效團建模工具如下。
表11 藥效團建模工具
藥效團建模中涉及的步驟(圖4)如下:

圖4 藥效團建模中涉及的步驟
選擇一組訓練配體:對于藥效團模型開發,請選擇結構多樣化的化合物組。分子列表應包括活性和非活性化合物,因為藥效團模型必須能夠區分具有和沒有生物活性的分子。
構象分析:為每種選定化合物創建一個低能量構象列表,其中可能包括生物活性構象。
分子疊加:疊加分子低能構象的所有可能組合。可以擬合集合中所有分子中相似的官能團(例如,苯基環或羧酸基團)。假定活性構象是導致最佳擬合的構象集合。
抽象:創建疊加分子的抽象表示。例如,疊加的苯基環在更概念的意義上可以稱為“芳香環”藥效團元素。
驗證:藥效團模型是一種假設,用于解釋與同一生物靶標結合的一組化合物的藥理作用。
1.2 藥物設計和藥物發現中的人工智能
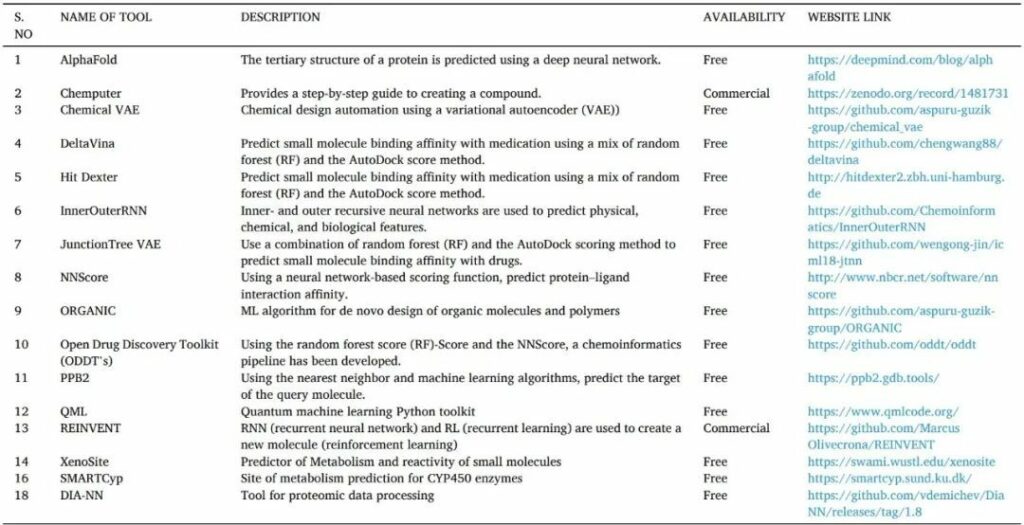
人工智能是通過機器或計算機對人類智能的模擬。它們通常通過訓練大量預先訓練的模型,分析相關性和模式的信息,然后使用這些模式進行預測來工作。人工智能可以識別hit和先導化合物,更快地驗證藥物靶標,并優化藥物結構設計。它還可以幫助靶向蛋白質的 3D 結構預測、蛋白質-蛋白質相互作用、藥物活性和從頭藥物設計(表13)。
表13 用于藥物發現的人工智能工具
2 結論
上述CADD技術被廣泛認為在所有情況下都遠非完美和無所不能。為了有效地采用當前的計算方法,必須克服相當大的限制。人工智能、機器學習和深度學習方法可以與基本的CADD程序一起使用,以提供更準確和準確的結果。
本文詳細解釋了到目前為止計算工具和技術是如何應用于藥物發現和開發的。還描述了藥物發現和開發過程中使用的當前工具和軟件列表。
- END -
我們有個生物/化學計算云平臺
集成多種CAE/CFD應用,大量任務多節點并行
應對短時間爆發性需求,連網即用
跑任務快,原來幾個月甚至幾年,現在只需幾小時
5分鐘快速上手,拖拉點選可視化界面,無需代碼
支持高級用戶直接在云端創建集群
掃碼免費試用,送200元體驗金,入股不虧~

更多電子書 歡迎掃碼關注小F(ID:imfastone)獲取

你也許想了解具體的落地場景:
這樣跑COMSOL,是不是就可以發Nature了
Auto-Scale這支仙女棒如何大幅提升Virtuoso仿真效率?
1分鐘告訴你用MOE模擬200000個分子要花多少錢
LS-DYNA求解效率深度測評 │ 六種規模,本地VS云端5種不同硬件配置
揭秘20000個VCS任務背后的“搬桌子”系列故事
155個GPU!多云場景下的Amber自由能計算
怎么把需要45天的突發性Fluent仿真計算縮短到4天之內?
5000核大規模OPC上云,效率提升53倍
提速2920倍!用AutoDock Vina對接2800萬個分子
從4天到1.75小時,如何讓Bladed仿真效率提升55倍?
從30天到17小時,如何讓HSPICE仿真效率提升42倍?
關于為應用定義的云平臺:
Uni-FEP on fastone|速石科技攜手深勢科技,助力創新藥物研發提速
【2021版】全球44家頂尖藥企AI輔助藥物研發行動白皮書
創新藥研發九死一生,CADD/AIDD是答案嗎?
這一屆科研計算人趕DDL紅寶書:學生篇
AI太笨了……暫時
幫助CXO解惑上云成本的迷思,看這篇就夠了
國內超算發展近40年,終于遇到了一個像樣的對手
花費4小時5500美元,速石科技躋身全球超算TOP500
【大白話】帶你一次搞懂速石科技三大產品:FCC、FCC-E、FCP