

本篇來說下CADD對藥物設(shè)計領(lǐng)域產(chǎn)生了重大影響。此外,CADD與人工智能(AI)、機(jī)器學(xué)習(xí)(ML)和深度學(xué)習(xí)(DL)技術(shù)相結(jié)合,處理大量生物數(shù)據(jù),減少了與藥物開發(fā)過程相關(guān)的時間和成本。
1.概述
計算機(jī)輔助藥物設(shè)計(CADD)結(jié)合了各種計算機(jī)工具,以識別和開發(fā)有前途的lead。CADD包括計算化學(xué)、分子建模、分子設(shè)計和合理的藥物設(shè)計。在當(dāng)今的大數(shù)據(jù)環(huán)境中,訪問大量數(shù)據(jù)并不能保證獲得適用的預(yù)測模型。為了預(yù)測治療效果和副作用,必須開發(fā)系統(tǒng)地解決大量、多維和稀疏數(shù)據(jù)源的技術(shù)。
1.1 計算機(jī)輔助藥物設(shè)計
CADD基于蛋白質(zhì)或配體的3D結(jié)構(gòu)的可用性,利用兩種不同的技術(shù)進(jìn)行藥物發(fā)現(xiàn):基于結(jié)構(gòu)的藥物設(shè)計(SBDD)和基于配體的藥物設(shè)計(LBDD)(圖1)。在某些情況下,兩種技術(shù)的整合在查找先導(dǎo)分子方面顯示出良好的準(zhǔn)確性。
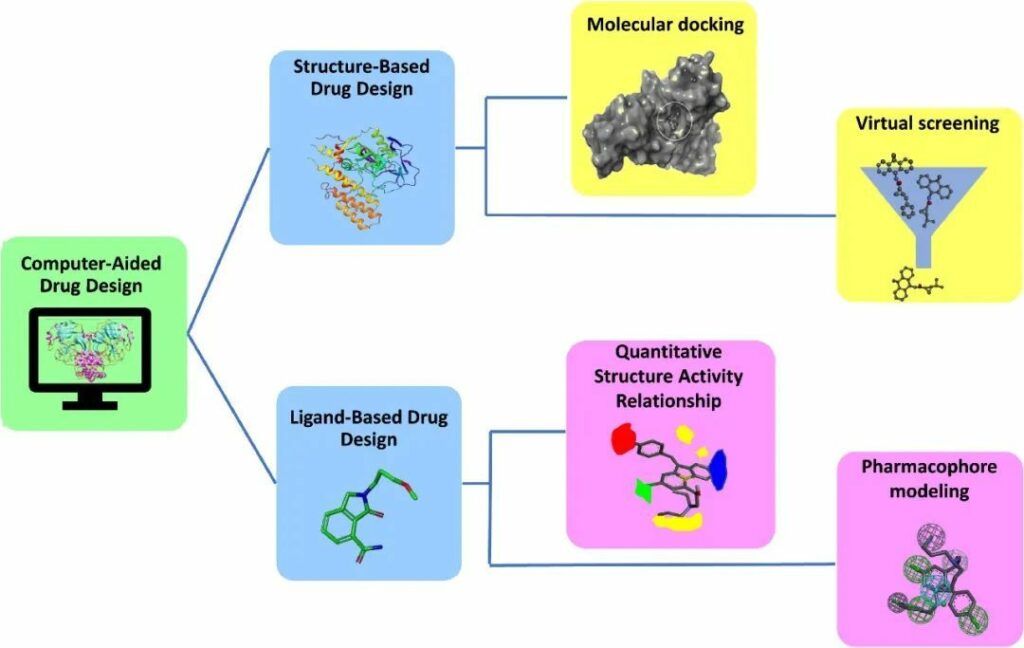
圖1 -CADD概覽
1.1.1 SBDD
基于結(jié)構(gòu)的藥物設(shè)計(SBDD):隨著越來越多生物分子的三維結(jié)構(gòu)的公開,SBDD在藥物發(fā)現(xiàn)和設(shè)計方面的新時代已經(jīng)開始。SBDD已成為制藥行業(yè)中生成和優(yōu)化配體的可能手段。靶標(biāo)的識別、結(jié)合位點的鑒定、分子對接、虛擬篩選和分子動力學(xué)是SBDD的基本步驟。
1.1.1.1靶標(biāo)準(zhǔn)備
準(zhǔn)備靶點大分子結(jié)構(gòu)是SBDD中最關(guān)鍵的一步。由于X射線和NMR結(jié)構(gòu)解析技術(shù)的快速發(fā)展,沉積在蛋白質(zhì)數(shù)據(jù)庫(PDB)中的蛋白質(zhì)的3D結(jié)構(gòu)很容易獲得。當(dāng)目標(biāo)蛋白的三維結(jié)構(gòu)不可用時,計算方法,如比較或同源建模(comparative or homology modeling)、threading和ab initio已經(jīng)能成功地從蛋白質(zhì)的序列中確定其結(jié)構(gòu)。
同源建模或比較建模:可以使用各種計算結(jié)構(gòu)預(yù)測技術(shù)(包括同源性建模)從其氨基酸序列推斷蛋白質(zhì)3D結(jié)構(gòu)(表1)。它被認(rèn)為是精度最高的計算結(jié)構(gòu)預(yù)測方法。其中有幾個簡單且易于遵循的步驟:尋找具有相似序列的結(jié)構(gòu)模板蛋白,對齊它們的序列,使用對齊的區(qū)域坐標(biāo),預(yù)測目標(biāo)缺失的原子坐標(biāo),模型構(gòu)建和細(xì)化。NCBI基本局部比對搜索工具(BLAST)是用于序列相似性搜索的最廣泛使用的生物信息學(xué)序列比對工具之一。
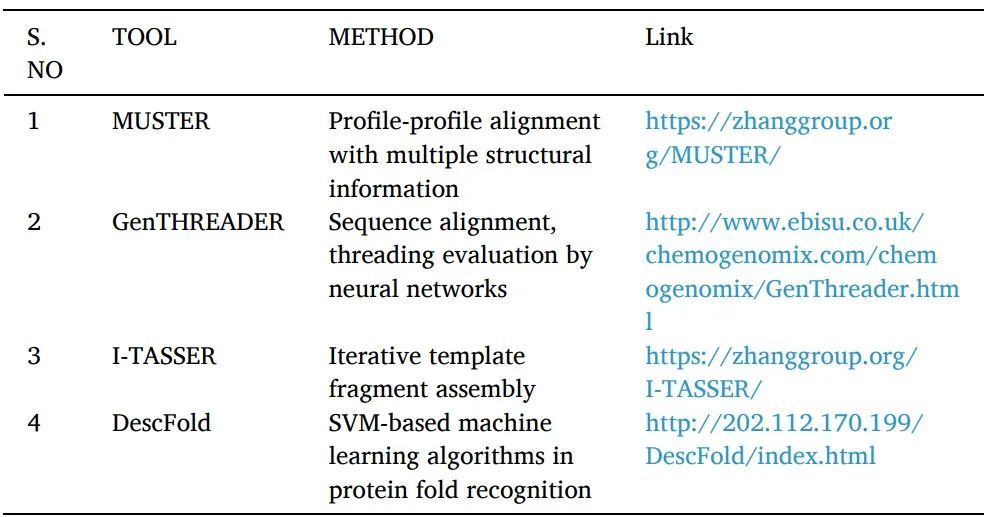
Fold recognition or threading:用于尋找具有可比折疊但沒有序列相似性的蛋白質(zhì)。一個已知的蛋白質(zhì)結(jié)構(gòu)的序列被感興趣的目標(biāo)的查詢序列所取代,對于該結(jié)構(gòu)是未知的。然后用各種評分系統(tǒng)對產(chǎn)生的"threaded"結(jié)構(gòu)進(jìn)行評估。對每個數(shù)據(jù)庫的經(jīng)驗確定的三維結(jié)構(gòu)重復(fù)這一過程,提供與查詢序列最匹配的結(jié)構(gòu)(表2)。它被運用于SBDD研究中。
表2 threading方法工具
從頭或從頭建模:當(dāng)結(jié)構(gòu)中沒有足夠的同質(zhì)性來進(jìn)行比較建模時,將執(zhí)行從頭或從頭建模(表3)。
表3 用于從頭建模的工具

1.1.1.2活性結(jié)合位點的鑒定和表征
藥物活性需要蛋白質(zhì)和配體的相互作用。只有找到高親和力結(jié)合位點才有可能。在基于結(jié)構(gòu)的藥物發(fā)現(xiàn)方法中開發(fā)新方法在很大程度上取決于識別靶蛋白上的可藥腔或口袋。
POCKET,SURFNET,Q-SITE FINDER,DoGSite Scorer server,CASTp,NSiteMatch,metapocket等工具是用于預(yù)測靶點蛋白結(jié)合位點的計算機(jī)工具。
找到結(jié)合位點后,使用諸如Epock,TRAPP和POVME等工具或服務(wù)器來確定結(jié)合口袋的體積(表4)。
1.1.1.3 分子對接
表4 結(jié)合位點預(yù)測工具
分子對接是一種用于確定配體分子在大分子靶標(biāo)結(jié)合位點中的構(gòu)象和取向(統(tǒng)稱為“位置”)的技術(shù)。
搜索算法用于生成姿勢,然后使用評分技術(shù)進(jìn)行排名。許多生物過程,如信號傳遞、細(xì)胞控制和其他大分子組裝,依賴于分子識別,如酶-底物、藥物-蛋白質(zhì)、藥物-核酸、蛋白質(zhì)-核酸和蛋白質(zhì)-蛋白質(zhì)相互作用。采樣和評分是蛋白質(zhì)-配體對接方法的兩個關(guān)鍵組成部分。
配體采樣和蛋白質(zhì)靈活性是采樣的兩個方面,指的是在蛋白質(zhì)結(jié)合位點附近創(chuàng)建可能的配體結(jié)合方向/構(gòu)象。評分使用物理方法或經(jīng)驗預(yù)測單個配體取向/構(gòu)象的結(jié)合緊密性。
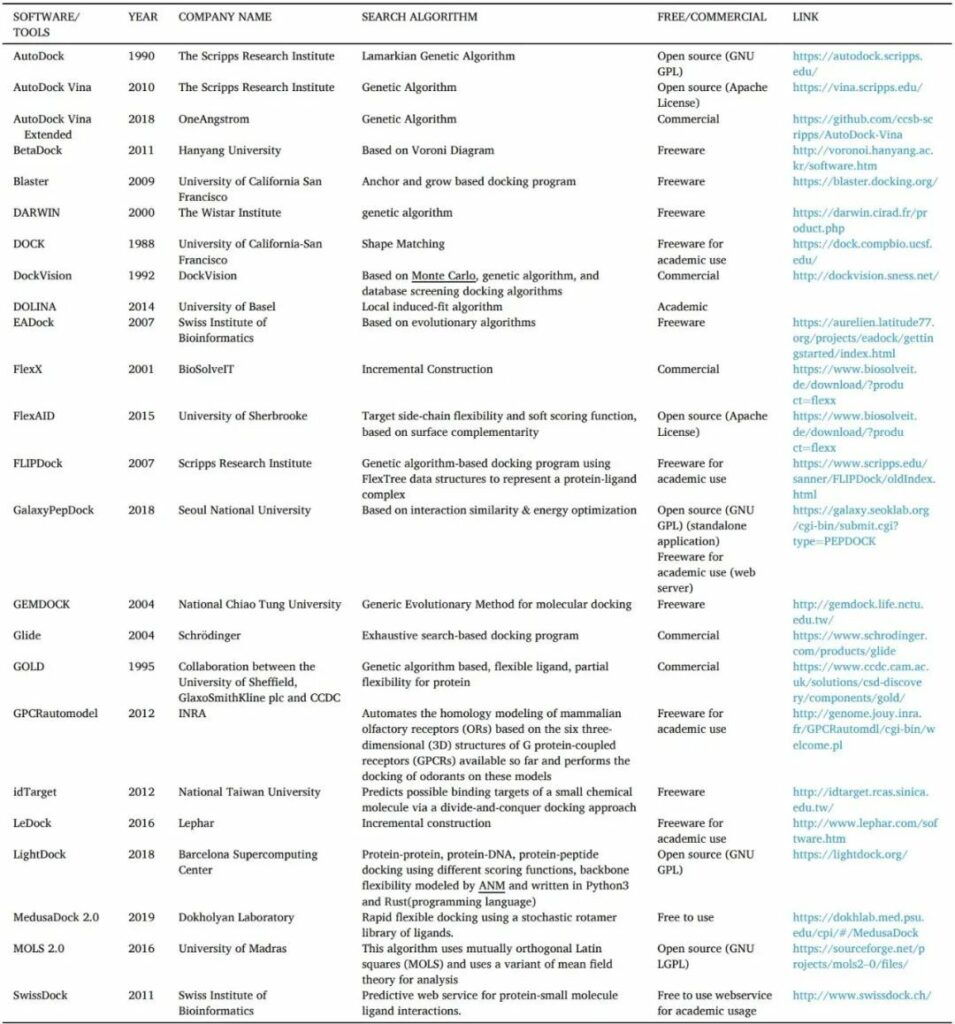
表5 對接工具/軟件的清單
蛋白質(zhì)-配體對接的分類方法有幾種。
根據(jù)蛋白質(zhì)靈活性,方法分為四類:
軟對接:在對接模擬中,它放松了原子間Vander Waals接觸,允許配體和蛋白質(zhì)之間略有重疊。
側(cè)鏈靈活性:早期的研究之一是Leach的配體對接方法,該方法使用旋轉(zhuǎn)體庫來結(jié)合離散的側(cè)鏈靈活性。從那時起,已經(jīng)提出了一系列新技術(shù),用于在配體對接中添加連續(xù)或離散的側(cè)鏈靈活性。
分子弛豫:第三種方法考慮蛋白質(zhì)的柔韌性,通過使用剛體對接將配體引入結(jié)合位點,然后松弛蛋白質(zhì)主鏈和附近的側(cè)鏈原子。初始剛體對接允許蛋白質(zhì)和插入的配體取向/構(gòu)象之間的原子沖突,以適應(yīng)蛋白質(zhì)構(gòu)象差異。使用蒙特卡羅(MC)模擬,分子動力學(xué)模擬或其他方法松弛或最小化復(fù)合物。
蛋白質(zhì)集合對接:添加蛋白質(zhì)柔韌性的最廣泛使用的方法涉及蛋白質(zhì)結(jié)構(gòu)的集合,以反映各種構(gòu)象變化。
根據(jù)配體采樣,方法分為兩類:
有兩種配體采樣算法:系統(tǒng)搜索和隨機(jī)算法(表6)。
表6使用隨機(jī)或系統(tǒng)搜索算法的軟件/工具
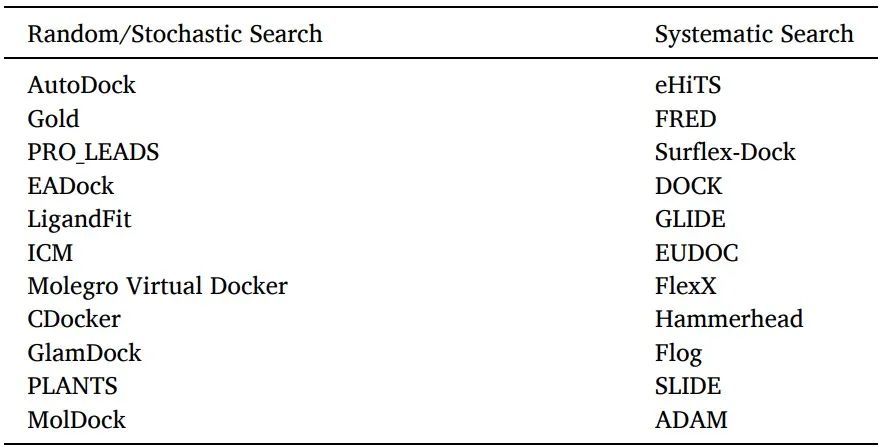
系統(tǒng)搜索:對于靈活的配體對接,通常使用系統(tǒng)搜索技術(shù),通過探索配體的所有自由度來創(chuàng)建所有潛在的配體結(jié)合構(gòu)象。
隨機(jī)算法:通過在構(gòu)象空間和配體的平移/旋轉(zhuǎn)空間的每一步對配體進(jìn)行隨機(jī)修改,在隨機(jī)算法中對配體結(jié)合取向和構(gòu)象進(jìn)行采樣。
根據(jù)評分功能,方法分為三類:
對接分?jǐn)?shù)是基于評估復(fù)合體能量親和力的評分函數(shù)的計算。這些評分函數(shù)可以在分子力學(xué)、經(jīng)驗數(shù)據(jù)、專業(yè)知識或共識庫上找到。共識評分(Consensus scoring)是一種通過組合幾種評分算法的結(jié)果來預(yù)測化合物對特定靶標(biāo)的結(jié)合親和力的方法。根據(jù)推導(dǎo)方法分為三個基本類別:基于力場、基于經(jīng)驗和基于知識的評分函數(shù)。
力場(FF)評分函數(shù):基于配體結(jié)合能分解為單個相互作用項,例如范德華(VDW)能量,靜電能,鍵拉伸/彎曲/扭轉(zhuǎn)能等,使用一組派生的力場參數(shù),例如AMBER或CHARMM力場。
經(jīng)驗評分函數(shù):配合物的結(jié)合能得分是通過將幾個加權(quán)經(jīng)驗?zāi)茼棧ㄈ鏥DW能,靜電能,氫鍵能,脫溶劑化項,熵項,疏水性項等)相加得出。
基于知識的評分函數(shù):直接從實驗確定的蛋白質(zhì)配體復(fù)合物中的結(jié)構(gòu)信息生成平均力的電位,由玻爾茲曼反比關(guān)系描述為基于知識的評分函數(shù)提供基礎(chǔ)。
1.1.1.4 虛擬篩選(VS)
在計算機(jī)中,從化學(xué)數(shù)據(jù)庫中選擇有希望的化合物的方法被稱為虛擬篩選,并且可以被認(rèn)為是實驗生物學(xué)評估方法的計算機(jī)化等效物,如高通量篩選(HTS)。
VS分為兩類:(i)基于結(jié)構(gòu)的虛擬篩選(SBVS)和(ii)基于配體的虛擬篩選(LBVS)(圖2)。

圖2 基于結(jié)構(gòu)和基于配體的虛擬篩選概述
基于結(jié)構(gòu)的虛擬篩選(SBVS):這是一種基于計算機(jī)的方法,用于在早期藥物開發(fā)項目中針對特定治療靶點搜索化合物庫中的新型生物活性化合物。SBVS中的化合物數(shù)據(jù)庫停靠在預(yù)定的靶標(biāo)結(jié)合位點。除了預(yù)測結(jié)合模式外,SBVS還為對接的分子分配排名。該評級可以用作選擇有前途的分子的唯一標(biāo)準(zhǔn),也可以與其他評估方法結(jié)合使用。進(jìn)行實驗以確定所研究分子靶標(biāo)上指示藥物的生物活性。SBVS包括四個步驟:(i)分子靶標(biāo)準(zhǔn)備(ii)化合物數(shù)據(jù)庫選擇(iii)分子對接和(iv)對接后分析。
表7 用于以圖形方式顯示SBVS和分子對接結(jié)果的程序
基于配體的虛擬篩選(LBVS):通過采用稱為基于配體的虛擬篩選的計算技術(shù),可以根據(jù)有效結(jié)合到靶標(biāo)的配體的信息生成靶蛋白的模型。之后,使用該模型預(yù)測新配體與靶標(biāo)結(jié)合的可能性。LBVS是唯一沒有靶蛋白3D結(jié)構(gòu)的方法。LBVS試圖使用已知的活性化學(xué)物質(zhì)作為輸入信息來識別具有相似屬性的結(jié)構(gòu)多樣化的分子。
1.1.1.5 分子動力學(xué)(MD)模擬
這種復(fù)雜的物理技術(shù)基于牛頓引導(dǎo)原子間相互作用的運動方程。它用于預(yù)測分子系統(tǒng)中每個原子相對于時間的位置。分子動力學(xué)(MD)模擬對于研究蛋白質(zhì)行為至關(guān)重要。在MD模擬中,化學(xué)鍵和鍵角使用簡單的虛擬彈簧描繪,而二面角則使用正弦函數(shù)處理。GPU 最初設(shè)計用于加速視頻游戲,現(xiàn)在正用于顯著加速分子動力學(xué)模擬。分子力學(xué)泊松-玻爾茲曼表面積(MM / PBSA),線性相互作用能(LIE)(和自由能擾動方法(FEP)是用于自由能計算的一些MD應(yīng)用,以關(guān)聯(lián)實驗和計算的小分子與蛋白質(zhì)的結(jié)合親和力。分子動力學(xué)模擬可以使用牛頓物理學(xué)和力場(如Amber或CHARMM OPLS,GROMOS和粗粒度力場(表8)計算構(gòu)象軌跡作為時間的函數(shù)。
表8 MD仿真中使用的力場列表

AMBER(具有能量細(xì)化的輔助模型構(gòu)建):AMBER是一組分子模擬程序和一組用于模擬生物分子的分子機(jī)械力場。Peter Kollman的小組在1970年代后期在加州大學(xué)舊金山分校開發(fā)了這種方法,以研究各種分子,如蛋白質(zhì),DNA,RNA,碳水化合物,有機(jī)分子,蛋白質(zhì)模擬物,脂質(zhì)和氟化芳香族氨基酸。為了施加分子對稱性,在全原子原子輔助模型構(gòu)建與能量細(xì)化(AMBER)系列力場中,部分電荷被分配具有靜電表面電位。
CHARMM(哈佛大學(xué)大分子力學(xué)化學(xué)):CHARMM程序最初由哈佛大學(xué)的Martin Karplus教授小組開發(fā),該小組協(xié)調(diào)經(jīng)驗力場參數(shù)化的努力。它具有針對各種分子參數(shù)化的特定力場。CHARMM力場中的部分電荷通常適合從頭計算的尺度能量。
OPLS(液體模擬的優(yōu)化電位):與實驗結(jié)果相比,OPLS力場在預(yù)測蒙特卡羅模擬獲得的結(jié)構(gòu)和熱力學(xué)特性方面表現(xiàn)出準(zhǔn)確性。
OPLS-AA力場,用于重現(xiàn)小分子的量子力學(xué)構(gòu)象能量分布。此外,它還從AMBER中獲得了幾個粘結(jié)參數(shù)。
GROMOS(格羅寧根分子模擬):GROMOS是一種用于研究生物分子系統(tǒng)的分子動力學(xué)的多用途計算機(jī)模擬工具。它還具有內(nèi)置力場,包括蛋白質(zhì)、核苷酸、糖和其他分子。它可用于模擬各種化學(xué)和物理系統(tǒng),包括玻璃、液晶、聚合物、晶體和生物分子溶液。
粗粒度力場(CG):CG力場通過減少模型中的自由度數(shù)來降低計算的計算成本,從而允許對更大的系統(tǒng)進(jìn)行更長時間的仿真。粗粒度(CG)模型有兩種常規(guī)方法:自下而上和自上而下。許多化合物的極性相和非極性相之間的自由能分配是CGMartini力場的基礎(chǔ)。Martini力場也是與原子模型密切合作開發(fā)的,特別是在束縛相互作用方面。
1.1.2 LBDD
在沒有關(guān)于受體的3D信息的情況下,可使用基于配體的藥物設(shè)計。該技術(shù)依賴于與感興趣的生物靶標(biāo)結(jié)合的分子知識。
與靶標(biāo)結(jié)合的已知配體的化學(xué)指紋圖譜用于分子相似性方法,以使用分子庫進(jìn)行篩選來鑒定具有相似指紋的化合物(表9)。配體相似性搜索方法是有效的,因為結(jié)構(gòu)相關(guān)的化合物具有相當(dāng)?shù)慕Y(jié)合特性。
表9 小分子數(shù)據(jù)庫

QSAR和藥效團(tuán)是LBDD的兩種方法。
1.1.2.1 QSAR
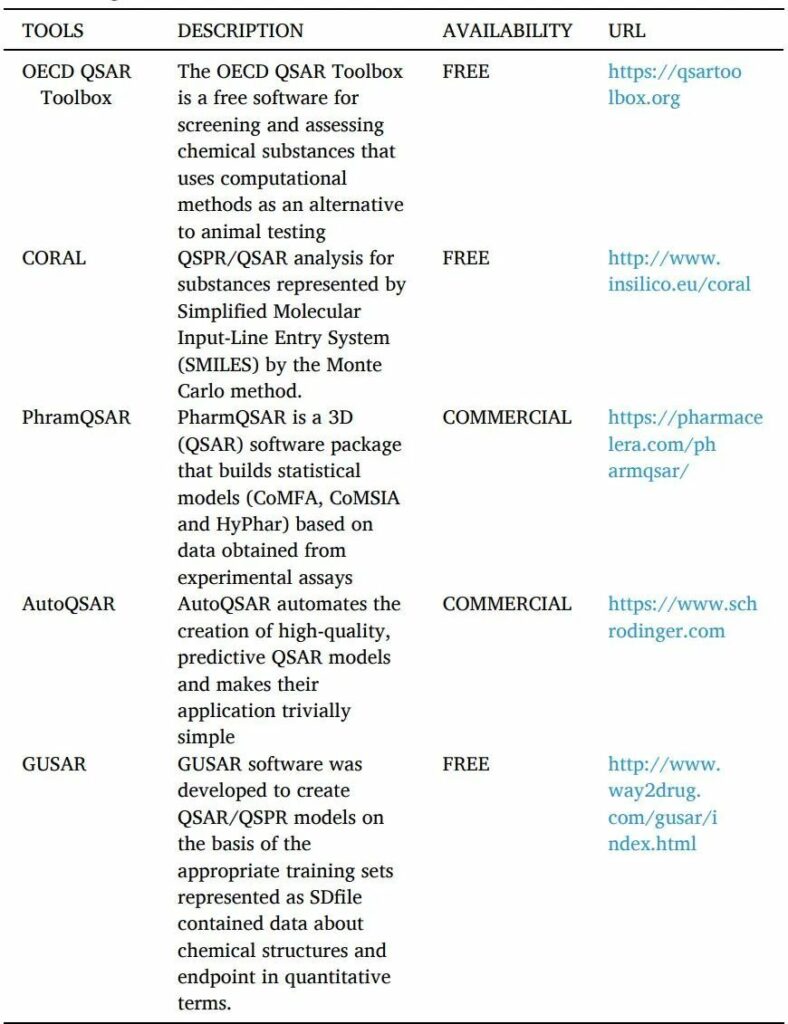
QSAR是一種計算機(jī)化的統(tǒng)計工具,用于解釋觀察到的由替換產(chǎn)生的結(jié)構(gòu)變化(表10)。這些模型用數(shù)學(xué)方法證明配體的結(jié)構(gòu)特性如何影響與之結(jié)合的靶點的活性反應(yīng)。可用于建立QSAR模型的分子參數(shù)可能包括電子、疏水、立體和亞結(jié)構(gòu)效應(yīng)。
QSAR的工具如下。
表10 QSAR工具
詳細(xì)參數(shù):
電子效果:電離常數(shù)、Σ取代基常數(shù)、分布常數(shù)、共振效應(yīng)、場效應(yīng)、分子軌道指數(shù)、原子/電子凈電荷、親核超離域性、親電超離域性、自由基超離域性、最低空分子軌道和最高占據(jù)分子軌道的能量、前沿原子-原子極化率、分子間庫侖相互作用能、由一組電荷在點(A)處產(chǎn)生的電場分子。
疏水參數(shù):分配系數(shù)、Pi取代基常數(shù)、液-液色譜中的Rm值、高壓液相色譜(HPLC)中的洗脫時間、溶解度、溶劑分配系數(shù)。
空間效應(yīng):分子內(nèi)空間位阻效應(yīng)、空間位阻取代基常數(shù)、超共軛校正、摩爾體積、摩爾折射率、MR 取代基常數(shù)、分子量、范德華半徑原子間距離。
子結(jié)構(gòu)效應(yīng):三維幾何碎片和分子性質(zhì)。
QSAR的步驟(圖3)如下:

制備用于QSAR實驗的分子:獲得一組在類似生物學(xué)測定中測試并顯示出廣泛作用的同屬配體。
訓(xùn)練集中描述符的選擇:識別并確定與化合物理化性質(zhì)相關(guān)的分子描述符。
計算訓(xùn)練集中描述符的值:將分子隨機(jī)分為兩組:訓(xùn)練集和測試集。使用訓(xùn)練集,識別并計算可以解釋描述符值與生物活性之間關(guān)系的相關(guān)系數(shù)。
內(nèi)部和外部驗證評估:使用測試集分子,評估統(tǒng)計方法的穩(wěn)定性。
1.1.2.2 藥效團(tuán)建模
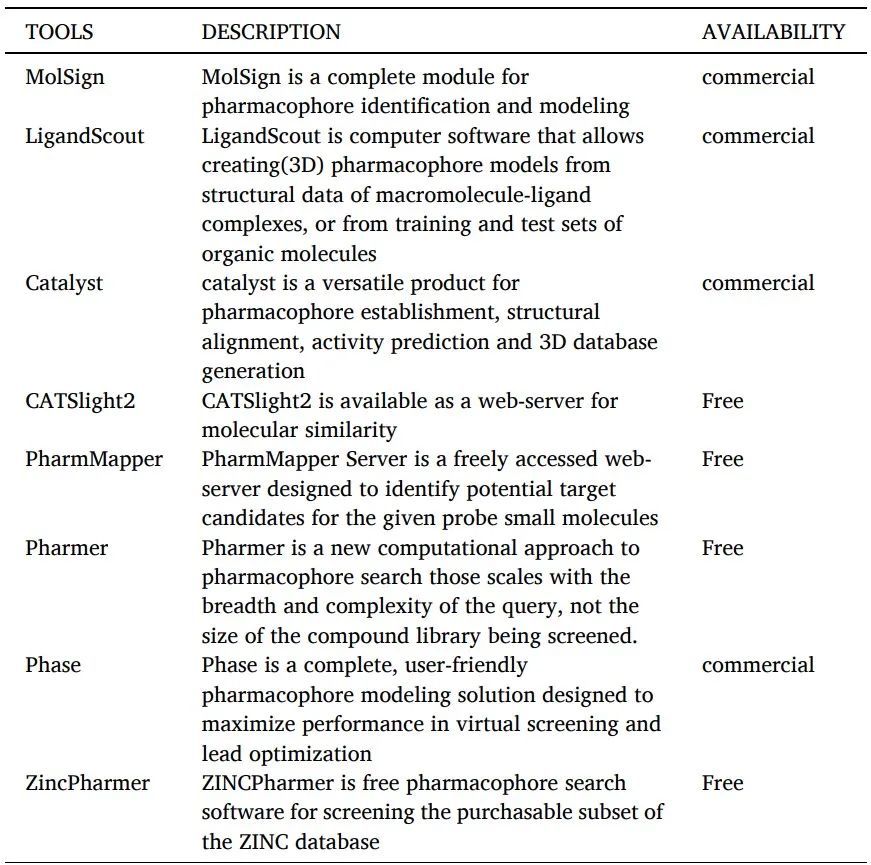
根據(jù)IUPAC的定義,藥效團(tuán)是“實現(xiàn)與給定生物靶標(biāo)的最佳超分子相互作用并觸發(fā)或預(yù)防其生物反應(yīng)所需的空間位阻和電子特性的集合”。藥效團(tuán)是對生物大分子識別配體所需結(jié)構(gòu)特性的抽象描述(表11)。
藥效團(tuán)建模工具如下。
表11 藥效團(tuán)建模工具
藥效團(tuán)建模中涉及的步驟(圖4)如下:

圖4 藥效團(tuán)建模中涉及的步驟
選擇一組訓(xùn)練配體:對于藥效團(tuán)模型開發(fā),請選擇結(jié)構(gòu)多樣化的化合物組。分子列表應(yīng)包括活性和非活性化合物,因為藥效團(tuán)模型必須能夠區(qū)分具有和沒有生物活性的分子。
構(gòu)象分析:為每種選定化合物創(chuàng)建一個低能量構(gòu)象列表,其中可能包括生物活性構(gòu)象。
分子疊加:疊加分子低能構(gòu)象的所有可能組合。可以擬合集合中所有分子中相似的官能團(tuán)(例如,苯基環(huán)或羧酸基團(tuán))。假定活性構(gòu)象是導(dǎo)致最佳擬合的構(gòu)象集合。
抽象:創(chuàng)建疊加分子的抽象表示。例如,疊加的苯基環(huán)在更概念的意義上可以稱為“芳香環(huán)”藥效團(tuán)元素。
驗證:藥效團(tuán)模型是一種假設(shè),用于解釋與同一生物靶標(biāo)結(jié)合的一組化合物的藥理作用。
1.2 藥物設(shè)計和藥物發(fā)現(xiàn)中的人工智能
人工智能是通過機(jī)器或計算機(jī)對人類智能的模擬。它們通常通過訓(xùn)練大量預(yù)先訓(xùn)練的模型,分析相關(guān)性和模式的信息,然后使用這些模式進(jìn)行預(yù)測來工作。人工智能可以識別hit和先導(dǎo)化合物,更快地驗證藥物靶標(biāo),并優(yōu)化藥物結(jié)構(gòu)設(shè)計。它還可以幫助靶向蛋白質(zhì)的 3D 結(jié)構(gòu)預(yù)測、蛋白質(zhì)-蛋白質(zhì)相互作用、藥物活性和從頭藥物設(shè)計(表13)。
表13 用于藥物發(fā)現(xiàn)的人工智能工具
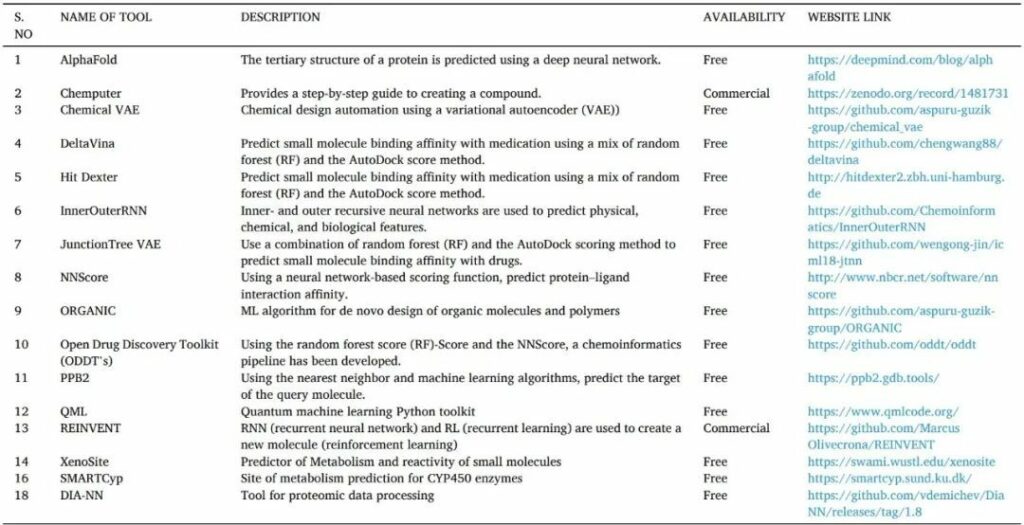
2 結(jié)論
上述CADD技術(shù)被廣泛認(rèn)為在所有情況下都遠(yuǎn)非完美和無所不能。為了有效地采用當(dāng)前的計算方法,必須克服相當(dāng)大的限制。人工智能、機(jī)器學(xué)習(xí)和深度學(xué)習(xí)方法可以與基本的CADD程序一起使用,以提供更準(zhǔn)確和準(zhǔn)確的結(jié)果。
本文詳細(xì)解釋了到目前為止計算工具和技術(shù)是如何應(yīng)用于藥物發(fā)現(xiàn)和開發(fā)的。還描述了藥物發(fā)現(xiàn)和開發(fā)過程中使用的當(dāng)前工具和軟件列表。
- END -
我們有個生物/化學(xué)計算云平臺
集成多種CAE/CFD應(yīng)用,大量任務(wù)多節(jié)點并行
應(yīng)對短時間爆發(fā)性需求,連網(wǎng)即用
跑任務(wù)快,原來幾個月甚至幾年,現(xiàn)在只需幾小時
5分鐘快速上手,拖拉點選可視化界面,無需代碼
支持高級用戶直接在云端創(chuàng)建集群
掃碼免費試用,送200元體驗金,入股不虧~

更多電子書 歡迎掃碼關(guān)注小F(ID:imfastone)獲取

你也許想了解具體的落地場景:
這樣跑COMSOL,是不是就可以發(fā)Nature了
Auto-Scale這支仙女棒如何大幅提升Virtuoso仿真效率?
1分鐘告訴你用MOE模擬200000個分子要花多少錢
LS-DYNA求解效率深度測評 │ 六種規(guī)模,本地VS云端5種不同硬件配置
揭秘20000個VCS任務(wù)背后的“搬桌子”系列故事
155個GPU!多云場景下的Amber自由能計算
怎么把需要45天的突發(fā)性Fluent仿真計算縮短到4天之內(nèi)?
5000核大規(guī)模OPC上云,效率提升53倍
提速2920倍!用AutoDock Vina對接2800萬個分子
從4天到1.75小時,如何讓Bladed仿真效率提升55倍?
從30天到17小時,如何讓HSPICE仿真效率提升42倍?
關(guān)于為應(yīng)用定義的云平臺:
Uni-FEP on fastone|速石科技攜手深勢科技,助力創(chuàng)新藥物研發(fā)提速
【2021版】全球44家頂尖藥企AI輔助藥物研發(fā)行動白皮書
創(chuàng)新藥研發(fā)九死一生,CADD/AIDD是答案嗎?
這一屆科研計算人趕DDL紅寶書:學(xué)生篇
AI太笨了……暫時
幫助CXO解惑上云成本的迷思,看這篇就夠了
國內(nèi)超算發(fā)展近40年,終于遇到了一個像樣的對手
花費4小時5500美元,速石科技躋身全球超算TOP500
【大白話】帶你一次搞懂速石科技三大產(chǎn)品:FCC、FCC-E、FCP






